Citrin deficiency (CD) is an inherited autosomal recessive metabolic condition that is also a urea cycle disorder caused by mutations in the SLC25A13 gene encoding for citrin. Citrin is the mitochondrial aspartate-glutamate carrier, a component of the malate-aspartate shuttle involved in moving reducing equivalents (NADH) from the cytosol into the mitochondria and for supplying aspartate to the cytosol. CD is a complex disorder with diverse phenotypes. The condition primarily affects the liver with several major pathways including glycolysis, gluconeogenesis, lipid metabolism, the TCA cycle, and ureagenesis being impacted [Saheki et al. 2020; Häberle and Rubio, 2022].
An emerging theory is that some of the underlying pathogenesis of citrin deficiency may be explained by a chronic energy deficit in the liver due to the inability of liver cells (hepatocytes) to efficiently utilize glucose and fatty acids as energy sources to produce ATP [Hayasaka, 2021; Hayasaka, 2023].
If you are a citrin deficiency patient, please click here for more information.
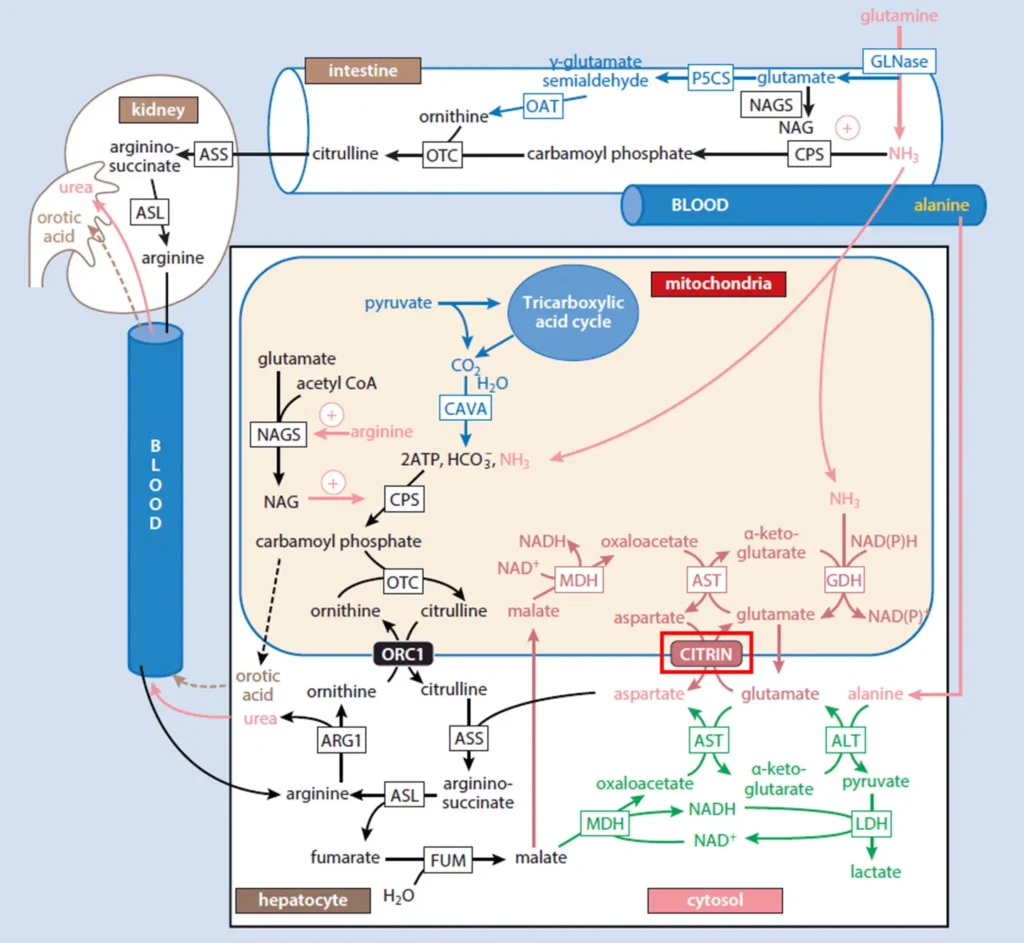
Figure 1. The role of citrin in the urea cycle. Citrin (red box) plays a key role in the urea cycle by transporting mitochondrial aspartate to the cytosol. Aspartate condenses with citrulline to form argininosuccinate, with the reaction being catalyzed by the enzyme argininosuccinate synthetase (ASS). Argininosuccinate then undergoes subsequent steps in the urea cycle pathway to produce urea, which is the end-product of the urea cycle. Urea is then excreted from the body via urine. Adapted from [Häberle and Rubio, 2022].
Please find the list of references here.