The diagnosis of citrin deficiency (CD) is based on clinical presentations and biochemical analysis (Table 1), with genetic analysis of the SLC25A13 gene being the gold standard. Patients suspected of CD should be referred to DNA testing for a confirmed diagnosis and if a genetic mutation in the gene is identified, a definite diagnosis of CD can be made. The following sections detail the specific diagnosis associated with each CD phenotype.
TABLE 1. Biochemical findings in citrin deficiency by phenotype [Adapted from Saheki & Song 2005; last updated 2017]
| Phenotype (Age) | Blood or Plasma Concentration of Ammonia (umol/L) | Plasma or Serum Concentration of Citrulline (C)# | Plasma or Serum Concentration of Arginine (A) (umol/L) | Plasma or Serum Threonine-to-Serine Ratio | Serum Concentration of Pancreatic Secretory Trypsin Inhibitor (PSTI)^ (ng/mL) |
|---|---|---|---|---|---|
| Control | 18-47* | 17-43* | 54-130* | 1.10 | 4.6-20* |
| NICCD (0-6 months) | 60 | 300 | 205 | 2.29 | 30 |
| FTTDCD | Normal or slightly elevated | Normal or slightly elevated | Typically normal | Unknown | Unknown |
| Adolescent and Adult Citrin Deficiency (AACD)* | 152 | 418 | 198 | 2.32 | 71 |
# Citrullinemia, which can be detected on newborn screening, is the earliest identifiable biochemical abnormality of NICCD [Tamamori et al 2004].
^ Because the serum PSTI concentration is high in some individuals with NICCD [Tamamori et al 2002] and also in individuals before the onset of AACD* [Tsuboi et al 2001], the measurement of serum PSTI concentration may be useful in the pre-symptomatic diagnosis of AACD*.
* Range
PHENOTYPES
Newborn screening (NBS) may pick up suspected cases if they show elevated citrulline, threonine, methionine, tyrosine, and phenylalanine (Figure 1) [Nakamura et al. 2019*]. Infants presenting with intrahepatic cholestasis, increased plasma citrulline without significant hyperammonemia (although mild/moderate elevations of ammonia are sometimes possible), normal or increased arginine without urinary orotic acid, high plasma levels of α-fetoprotein, and/or increased galactose in blood and urine are strongly suspected to have NICCD [Häberle & Rubio 2022]. NICCD patients may also exhibit higher levels of serum gamma-glutamyl transferase (GGT, or GTP) [Saheki & Song 2005; last updated 2017, Nakamura et al. 2019*].
Patients identified through newborn screening with increased blood citrulline should first exclude the possibility of argininosuccinate synthetase (ASS) deficiency. In severe ASS deficiency, plasma ammonia, glutamine, and urinary orotic acid levels are significantly elevated but are not in NICCD. On the other hand, arginine levels are low or normal in ASS deficiency while elevated in NICCD [Häberle & Rubio 2022].
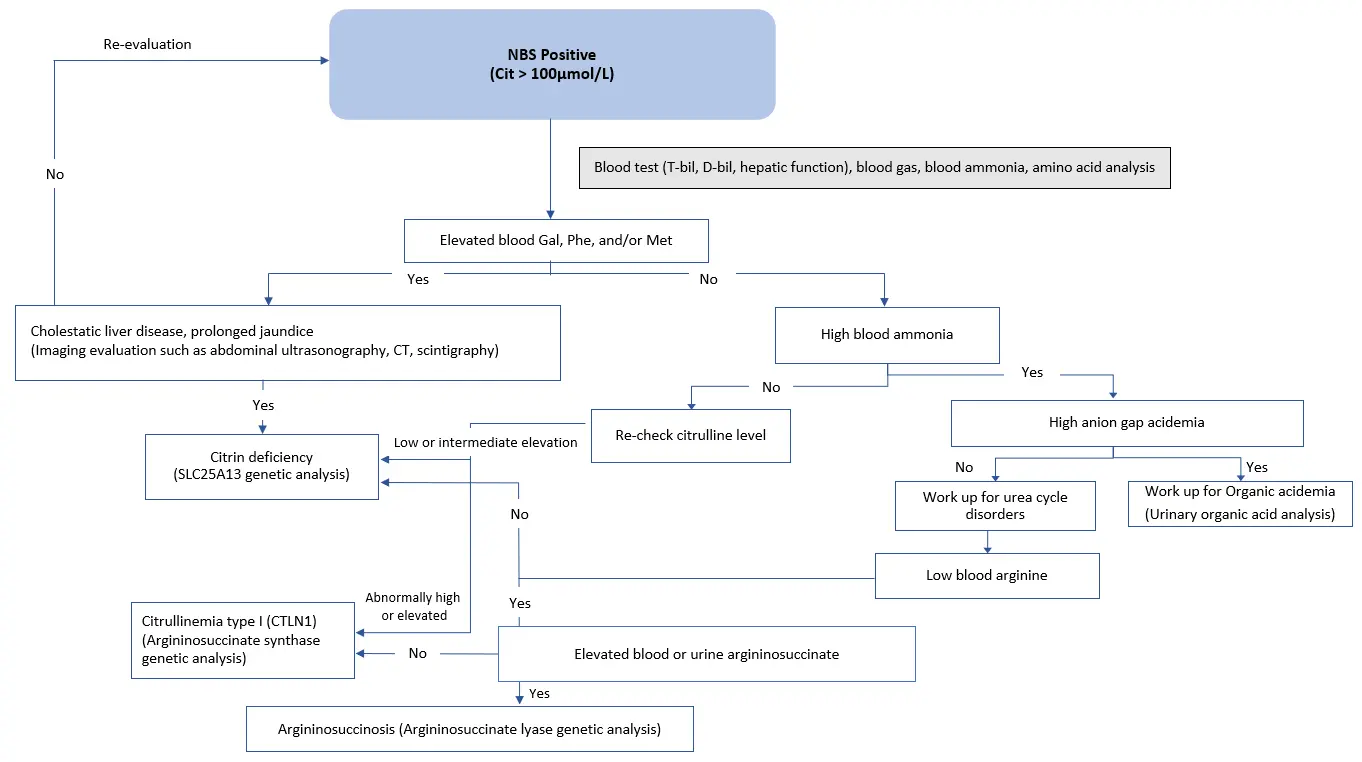
Figure 1. NBS flow chart for newborns identified with elevated citrulline levels. Differential diagnosis is performed if newborns are identified with elevated citrulline levels by referring to the following flow chart. A definitive diagnosis is achieved by genetic analysis of the SLC25A13 gene. [Adapted and translated from Nakamura et al. 2019*. Original text available in Japanese only]
Abbreviations: Cit = citrulline; D-bil = direct bilirubin; Gal = galactose; Met = methionine; NBS = newborn screening; Phe = phenylalanine; T-bil = total bilirubin
Unless NICCD has been previously confirmed, the specific diagnosis of patients in the adaptation period may be difficult to ascertain. A specific dietary preference towards foods that are high in protein and fat while avoiding carbohydrates and sugars, however, may be indicative of citrin deficiency. High AST (aspartate aminotransferase), ALT (alanine aminotransferase), abdominal pain, fatty liver, and hypoglycemia especially during infections may also be indicative of the condition. FTTDCD may be suspected if citrullinemia, hypoglycemia, dyslipidemia, and high lactate/pyruvate ratios are also observed [Nakamura et al 2019*, Häberle & Rubio 2022] (Figure 2).
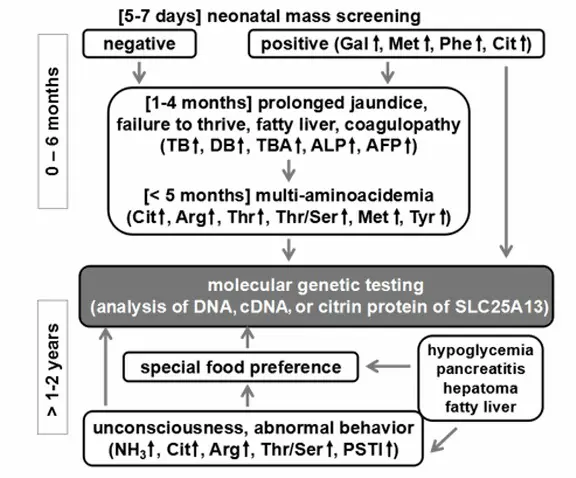
Figure 2. Flow chart for diagnosis of citrin deficiency in neonates and children (post-NICCD). Note that food preferences (e.g., strong liking for protein and fats, and aversion to carbohydrates and sugars) are important in the diagnosis of citrin deficiency. This applies not only in patients suspected of AACD*, but also in cases of growth retardation, hypoglycemia, pancreatitis, and hypertriglyceridemia in both children and adults. [Adapted from Saheki & Song 2005; last updated 2017]
Abbreviations: AFP = α-fetoprotein; ALP = alkaline phosphatase; DB = direct bilirubin; PSTI = pancreatic secretory trypsin inhibitor; TB = total bilirubin; TBA = total bile acids
Conscious disturbances, elevated AST, ALT, GGT, arginine, ammonia, and citrulline levels are common clinical presentations. Combined with a distinct food preference characteristic to citrin deficiency patients, this strongly indicates AACD* [Saheki & Song 2005; last updated 2017, Nakamura et al. 2019*, Häberle & Rubio 2022]. AACD* may be differentiated from ASS deficiency if plasma levels of glutamine are not significantly elevated with an absence of urinary orotic acid and normal or slightly elevated arginine levels [Häberle & Rubio 2022]. Patients admitted with conscious disturbances and hyperammonemia should refer to the diagnostic algorithm below (Figure 3) to rule out the possibility of other urea cycle disorders.
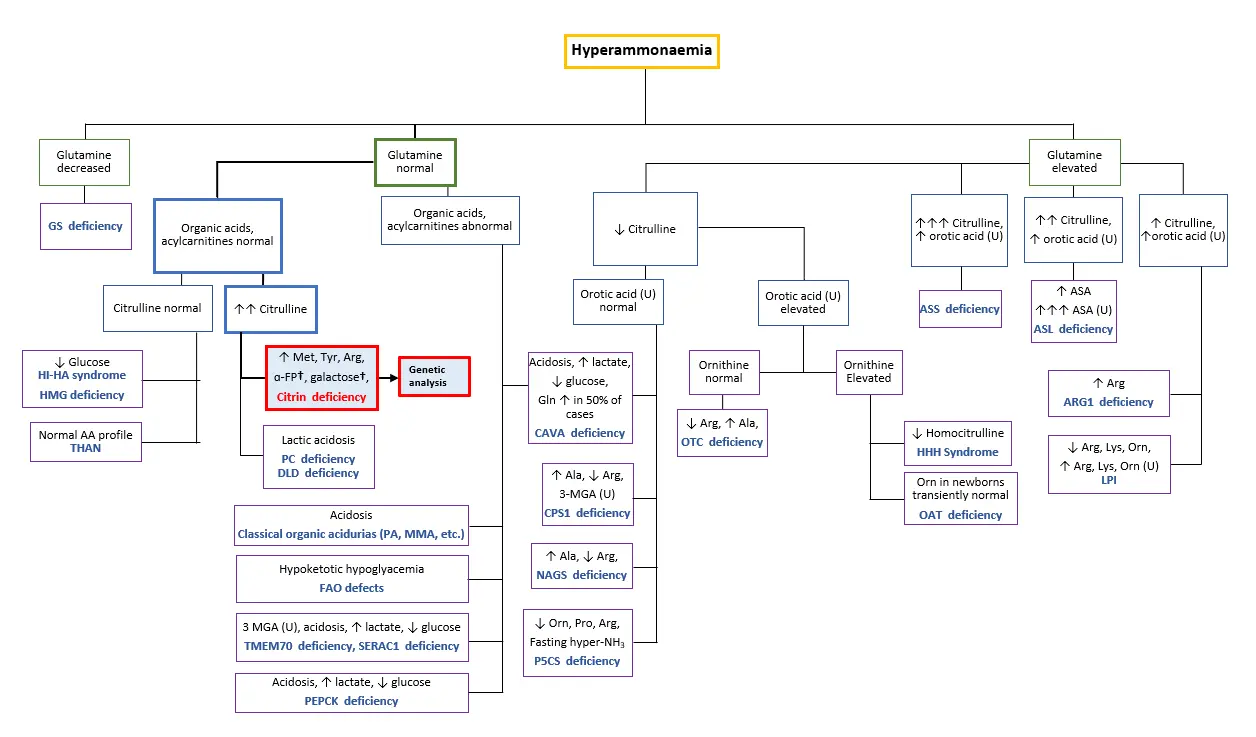
Figure 3. Diagnostic algorithm to differentiate and diagnose possible citrin deficiency patients admitted with hyperammonemia. †Elevated α-FP and galactose only in infants. [Adapted from Häberle & Rubio 2022]
Abbreviations: α-FP alpha-fetoprotein, Ala alanine, ASA argininosuccinic acid, ASL argininosuccinate lyase, Arg arginine, ARG1 arginase 1, ASS argininosuccinate synthetase, CAVA carbonic anhydrase Va, CPS carbamoyl phosphate synthetase, DLD dihydrolipoamide dehydrogenase, FAO fatty acid oxidation, Gln glutamine, GS glutamine synthetase, HHH hyperornithinaemia-hyperammonaemia-homocitrullinuria, HI-HA hyperinsulinism-hyperammonaemia, HMG 3-hydroxy-3-methylglutaryl-CoA lyase, LPI lysinuric protein intolerance, Met methionine, 3-MGA 3-methylglutaconic aciduria, MMA methylmalonic aciduria, NAGS N-acetylglutamate synthase, OAT ornithine aminotransferase, Orn ornithine, OTC ornithine transcarbamylase, PA propionic acidaemia, PC pyruvate carboxylase, P5CS Δ1-pyrroline-5-carboxylate synthetase, Pro proline, SERAC1 serine active site-containing protein 1, THAN transient hyperammonaemia of the newborn, TMEM70 transmembrane protein 70, Tyr tyrosine, U urine
*Previously known as Citrullinemia type 2 (CTLN2)
Please find the list of references here.